PRESENTACIÓN DE CASOS
Ataxia espinocerebelosa tipo 7. A propósito de un caso
Spinocerebellar ataxia type 7. About a case
Harold Cedeño Escalona 1* http://orcid.org/0000-0001-7772-4726
Carlos Alejandro de la Rosa Cruz 1 http://orcid.org/0000-0002-3450-4691
Maryela Landa Muñiz 2 http://orcid.org/0000-0001-5158-4059
1 Universidad de Ciencias Médicas de Matanzas. Matanzas, Cuba.
2 Centro Provincial De Genética Médica. Matanzas, Cuba.
*Autor para la correspondencia: harold.cedeno@nauta.cu
Recibido: 17/01/2023
Aceptado: 20/11/2023
RESUMEN
Introducción: la ataxia espinocerebelosa tipo 7 es una enfermedad neurodegenerativa de origen genético con una baja prevalencia entre la población. Se caracteriza por pérdida visual asociada a la presencia de ataxia. Su herencia es autosómica dominante.
Objetivo: describir una familia con diagnóstico clínico y molecular de dicha enfermedad y sus características distintivas.
Presentación del caso: el caso presentado consistió en una familia portadora de la mutación para la ataxia espinocerebelosa tipo 7. La enfermedad apareció de novo hace tres generaciones y se transmitió a la siguiente generación afectándose una de las tres hermanas que transmitió el gen mutado a sus dos hijos y uno de ellos, a la descendencia.
Conclusiones: este caso permitió observar tanto las características de la enfermedad, como el patrón de herencia, también su aparición en la edad adulta y el progresivo avance. Es una muestra de la importancia del análisis clínico minucioso en conjunto con las pruebas genéticas para lograr un diagnóstico definitivo.
Palabras clave: Ataxia; Ataxia Cerebelosa Autosómica Dominante; Ataxia Espinocerebelosa 7; Ataxias Hereditarias.
ABSTRACT
Introduction: spinocerebellar ataxia type 7 is a neurodegenerative disease of genetic origin with a low prevalence among the population. It is characterized by visual loss associated with the presence of ataxia. Its inheritance is autosomal dominant.
Objective: to describe a family with a clinical and molecular diagnosis of said disease and its distinctive characteristics.
Case presentation: the case presented consisted of a family carrying the mutation for spinocerebellar ataxia type 7. The disease appeared de novo three generations ago and was transmitted to the next generation, affecting one of the three sisters who transmitted the mutated gene to their children two children and one of them, to the offspring.
Conclusions: this case allowed us to observe both the characteristics of the disease, the inheritance pattern, its appearance in adulthood and the progressive progression. It is an example of the importance of thorough clinical analysis in conjunction with genetic testing to achieve a definitive diagnosis.
Keywords: Ataxia; Autosomal Dominant Cerebellar Ataxia; Spinocerebellar Ataxia 7; Hereditary Ataxias.
INTRODUCCIÓN
La ataxia espinocerebelosa tipo 7 (SCA7) constituye una enfermedad genética y neurodegenerativa que puede aparecer entre los 6 meses y los 60 años de edad. (1)
Su prevalencia estimada a nivel mundial es inferior a 1 por cada 100000 habitantes y se cree que es la responsable de entre el 2 % y el 4 % de todas las formas de ataxia espinocerebelosa. En algunas poblaciones, como la escandinava o la sudafricana se ha descrito una prevalencia más alta. En América Latina, México es el país con mayor prevalencia. (1, 2) En Cuba solamente se tiene registro del estudio de 2 familias con este tipo de ataxia espinocerebelosa. (3)
La SCA7 se produce por la repetición anormal en el gen ATXN7 del trinucleótido CAG, este codifica para una poliglutamina y se localiza en cromosoma 3p12-p21.1. (4) Tras el examen genético molecular se consideran alelos normales si el número de repeticiones está entre 7 y 27, de 28 a 33 constituyen alelos inestables; alelos con 34 a 36 se considera penetrancia incompleta con manifestaciones tardías y leves, y de 37-460 repeticiones es una mutación de penetrancia completa por lo que presentará una sintomatología clásica. (5)
Se transmite con un patrón autosómico dominante, en la que se puede observar un fenómeno de anticipación genética con la aparición más precoz de la sintomatología en las generaciones siguientes por un aumento en el número de repeticiones del trinucleótido CAG que ocurre fundamentalmente durante la gametogénesis. (6)
El cuadro clínico de la ataxia espinocerebelosa tipo 7 consiste en una ataxia cerebelosa progresiva con dismetría, disfagia, disartria, hiperreflexia y disdiadococinesia, procedida por una coordinación pobre. Se distingue de otros tipos de ataxia por la presencia de manifestaciones visuales relacionadas con la visión de colores y la agudeza visual con fotofobia, cambios pigmentarios, oftalmoplejía y degeneración de conos y bastones que conducen a la ceguera. Estas pueden preceder a las manifestaciones cerebelosas hasta en 9 años. (2,7)
Dada las características de la ataxia espinocerebelosa tipo 7 que la convierten en una enfermedad rara por su baja prevalencia y su expresión genotípica y fenotípica, el objetivo de esta presentación de caso es describir una familia con diagnóstico clínico y molecular de dicha enfermedad y sus características distintivas.
PRESENTACIÓN DEL CASO
En el año 2010 se le realizó el estudio genético de una familia donde se determinó la presencia de ataxia espinocerebelosa tipo 7 que se muestra en el árbol genealógico a continuación (figura. 1):
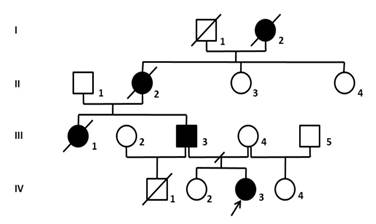
Fig. 1: Árbol genealógico de la familia objeto de estudio.
La paciente IV-3 acudió a consulta con 16 años de edad refiriendo inestabilidad para caminar y preocupada porque otros miembros de su familia comenzaron presentando características similares a ella y que se fueron agravando con el paso del tiempo hasta que murieron. Explica que en su familia la primera persona en padecer de la enfermedad fue su bisabuela que murió muchos años atrás. Esta tuvo 3 hijas de las cuales una resultó afectada, la abuela del propósito (II-2) que también está fallecida.
Su tía paterna (III-1) debutó con la enfermedad a los 15 años mostrando marcha atáxica, hipertonía espástica en miembros inferiores, hipotonía en miembros superiores, Babinski en pie izquierdo, temblor intencional y nistagmo horizontal, se le diagnosticó ataxia espinocerebelosa, pero nunca se le realizaron estudios genéticos y falleció a los 22 años.
El padre del propósito (III-3) que debutó a los 20 años, presenta también los síntomas y signos de la enfermedad en el momento del estudio, marcha atáxica y una ligera hipotonía de miembros superiores, actualmente, con 48 años, presenta ataxia y, además, una retinosis pigmentaria estadio 4 mientras que su madre no la padece. El propósito tuvo 3 hermanos.
Su hermano paterno (IV-1) se desconoce si estaba sano o afectado porque falleció siendo un neonato. Su hermana, del mismo padre y madre, (IV-2) tiene 31 años actualmente y es aparentemente sana. Además, tuvo una hermana materna que tampoco manifiesta síntomas.
Al examen físico presentaba marcha atáxica con tono y fuerza muscular conservada, maniobras de Romberg simple y sensibilizados positivos, no Babinski y no nistagmo. Se le realizó fondo de ojo al propósito que arrojo datos clínicos para el diagnóstico de una retinosis pigmentaria asociada. En el caso de su tía, se observaron las papilas pálidas, vasos finos y algunos pigmentos en el polo posterior correspondiendo también con esta patología.
Ante la descripción de las características clínicas, así como del patrón de herencia autosómica dominante con el que se corresponde, se sospecha que los familiares puedan padecer de ataxia espinocerebelosa y se remite a consulta genética para que se realicen pruebas que corroboren o descarten el diagnóstico.
Se realizaron las siguientes pruebas adicionales:
- Análisis molecular del ADN al propósito (IV-3) demostró la presencia de: Un alelo normal y un alelo expandido para el gen SCA 7, por lo que es heterocigótica con un alelo mutado (Figura. 2).
- Análisis molecular del ADN a su padre (III-3) demostró la presencia de: Un alelo normal y un alelo expandido para el gen SCA 7, por lo que es heterocigótica con un alelo mutado (Figura. 3).

Fig. 2: Informe de los Resultados del Estudio Molecular del individuo IV-3. Fuente: Historia clínica

Fig. 3: Informe de los Resultados del Estudio Molecular del individuo III-3. Fuente: Historia clínica
A la tía fallecida (III-1) se le realizaron otros estudios que se recogieron a través de su historia clínica como:
- Potenciales evocados auditivos de tronco cerebral (PEATC): Abolición de onda D en el oído izquierdo por probable lesión mesencefálica.
- Potencial evocado visual (PEV): No hubo respuesta.
- Potencial evocado somatosensorial (PESS): Lesión en tallo (MSD, MSI).
- TAC: Malformaciones vasculares o astrocitarias del cerebelo.
Mediante las pruebas genéticas se corroboró la presencia de ataxia espinocerebelosa tipo 7 (SCA 7) en la familia y se le informó a la paciente IV-3 que portaba el gen mutado. Se le explicó que existe una posibilidad del 50 % de que su descendencia herede la enfermedad y se le aconsejó realizar un test prenatal en el caso de quedar embarazada.
Luego del diagnóstico la paciente propósito continuó con seguimiento por Neurología y Oftalmología, sin tratamiento curativo al no contarse con una terapéutica efectiva para esta enfermedad. Continuó con una evolución clínica desfavorable debido al progreso de las manifestaciones clínicas, hasta hace 3 años que la paciente fallece con 26 años por complicaciones relacionadas con esta patología.
DISCUSIÓN DEL CASO
La ataxia espinocerebelosa tipo 7, en sí misma, es sumamente rara e interesante, la posibilidad de estudiar el caso de una familia afectada por esta enfermedad resulta una oportunidad única dada su baja prevalencia, existiendo registro de solo 2 familias en nuestro país. (3, 8, 9) Constituye, además, dentro de las enfermedades poliglutamínicas, una de las de más baja incidencia. (2)
Otra característica interesante es su origen, ya que no se trata de la típica mutación donde ocurre un cambio, se omite o se agrega alguna base nitrogenada, esta es una mutación dinámica donde el gen “crece” de una generación a la siguiente hasta producir una proteína defectuosa. (2, 10, 11)
La enfermedad presenta, además, una serie de manifestaciones neuropsicológicas bastante peculiares que la distinguen del resto de enfermedades neurodegenerativas, aparte de presentar ataxia asociado a degeneración visual se le sumó la afectación auditiva identificada por los potenciales evocados descrito en este tipo de ataxia. (9, 10, 12)
De esta manera se puede diferenciar de otros tipos de ataxia que, aunque comparten el cuadro clínico correspondiente al síndrome cerebeloso, se distinguen por otras manifestaciones asociadas. En la SCA1 se acompaña de polineuropatía, en la SCA2 existe enlentecimiento sacádico y polineuropatía sensitiva, la SCA3 presenta nistagmos y distonía, la SCA 6 solo se caracteriza por el síndrome cerebeloso. Otras posibilidades que podemos descartar en este caso por la demencia y epilepsia acompañante sería la SCA17. (2)
Además de las particularidades de la enfermedad en sí misma, en este caso se presentan varias características inusuales. Se observa la presencia de una nueva mutación, es decir que esta familia no heredó la enfermedad muchas generaciones atrás por ascendencia escandinava o sudafricana, como sucede en la mayoría de los casos, sino que la enfermedad surgió solamente tres generaciones atrás, diferente a como ocurre con otros tipos de ataxia de mayor prevalencia en Cuba. (2)
Algo significativo en la herencia de esta enfermedad que la distinguen del resto de los casos descritos es que, a pesar de tener un patrón de herencia autosómico dominante donde no están involucrados los cromosomas sexuales sino los autosómicos y, por ende, no debe estar influido por el sexo, se observa que el sexo femenino presenta el cuadro clínico de la enfermedad más grave y letal mientras que en el sexo masculino es más leve. (9,10,13)
Para descartar que esto se deba al fenómeno de la anticipación antes descrito, se deben analizar los individuos III-1 y III-3 que pertenecen a la misma generación y es su madre quien portaba la mutación. La tía del propósito (III-1) presentaba síntomas de ataxia cerebelosa más severo con un debut a una edad más temprana en la enfermedad, así como la muerte en correspondencia con el padre del paciente objeto de estudio (III-3). Esto sugiere que, aunque sea una enfermedad con un patrón de herencia autosómico dominante y no ligado al sexo parece estar influido por el mismo.
Tanto la ataxia espinocerebelosa como este caso particular tienen utilidad didáctica, ya que permite observar las clásicas características de la herencia autosómica dominante, además de fenómenos que interfieren en este tipo de herencia como el fenómeno de anticipación. (10) Al incluir estudios genéticos precisos se pueden observar las características del gen.
Como se ha observado en este caso, el pronóstico para esta enfermedad es desfavorable con una progresión de las manifestaciones atáxicas y visuales que conllevan a la muerte alrededor de 10 años tras el diagnóstico. (2) No existe un tratamiento médico efectivo aunque a nivel mundial se desarrollan ensayos clínicos aun en fases tempranas para limitar el avance de la enfermedad, entre ellos el interferón-β. Hasta el momento este se basa en la neurorehabilitación. (2, 10)
CONCLUSIONES
Este caso permitió observar tanto las características de la ataxia espinocerebelosa tipo 7 como su patrón de herencia que incluso pudiera estar influenciado por el sexo, su aparición en la adolescencia y edad adulta con el fenómeno de anticipación y su progresivo avance. Es una muestra de la importancia del análisis clínico minucioso para llegar a un diagnóstico presuntivo y de las pruebas genéticas para un diagnóstico definitivo.
REFERENCIAS BIBLIOGRÁFICAS
1. Beltran-Parrazal, Luis. La ataxia SCA7: un problema genético. En: Mario Caba, Rossana C. Zepeda, Enrique Meza, Claudia Juárez Portilla, editores. Avances en la investigación biomédica en el Estado de Veracruz. Primera edición. México: Centro de Investigaciones Biomédicas, Universidad Veracruzana; 2015. p. 313-321. Disponible en: https://www.researchgate.net/publication/299509479
2. Velázquez Pérez LC, Rodríguez Labrada R, Vázquez Mojena Y. Enfermedades poliglutamínicas. Diagnóstico clínico-genético y tratamiento. La Habana: Editorial Ciencias Médicas; 2019
3. González Zaldívar Y, Vázquez Mojena Y, Velázquez Pérez LC, Rodríguez Labrada R, Calnales Ochoa N, et al. Familias cubanas con ataxia espinocerebelosa tipo 7. Movement Disorders. 2013 [citado 06/01/2023]; 28:S247. Disponible en: https://www.researchgate.net/publication/256442054_Cuban_families_with_spinocerebellar_ataxia_type_7
4. Rojas JI., Romano M, Patrucco L, Zurrú MC, Igarreta P, Cristiano Edgardo. Ataxia espinocerebelosa 7: Investigación clínica y genética en una familia argentina. Medicina (B. Aires) [Internet]. 2007 [citado 06/01/2023]; 67(2):147-150. Disponible en: http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S0025-76802007000200007&lng=es
5. La Spada AR. Ataxia Spinocerebelosa Tipo 7. 27 de agosto de 1998 [Actualizado 23 de julio 2020]. En: Adam MP, Everman DB, Mirzaa GM, et al., Editores. GeneReviews [Internet]. Seattle (WA): Universidad de Washington, Seattle; 1993-2023. [citado 06/01/2023]. Disponible en: http://www.ncbi,nlm.nih.gov/books/NBK1256
6. Arnalich-Montiel F, Rebolleda G, Muñoz-Negrete FJ. SCA-7: Distrofia de conos-bastones en el seno de una ataxia hereditaria. Arch Soc Esp Oftalmol [Internet]. 2005 [citado 06/01/2023]; 80(11):679-682. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0365-66912005001100012&lng=es
7. Valdez Vargas, C. Perfil de expresión de miRNAs circulantes en pacientes con ataxia espinocerebelosa tipo 7 (SCA7). [Master’s thesis]. Ciudad de México: Cinvestav; 2019. 99 p. [citado 06/01/2023]. Disponible en: https://repositorio.cinvestav.mx/handle/cinvestav/2469
8. Dulski J, Hanna Al-Shaikh R, Prudencio M, Petrucelli L, Sulek A, Bernatowicz K, et al. First families with spinocerebellar ataxia type 7 in Poland. Polish Journal of Neurology and Neurosurgery. 2023 [citado 27/09/2023]; 57(3):310–313. Disponible en: https://journals.viamedica.pl/neurologia_neurochirurgia_polska/article/download/PJNNS.a2023.0037/71911
9. Velázquez-Pérez D. Ataxias hereditarias y enfermedad de Alzheimer familiar en América Latina: Contribución del proyecto cubano de ataxias. Anales de la Academia de Ciencias de Cuba [Internet]. 2018 [citado 27/09/2023]; 8(1). Disponible en: https://revistaccuba.sld.cu/index.php/revacc/article/view/437
10. Guevara López JC. Viviendo entre barrancas: un estudio bioantropológico de la ataxia espinocerebelar tipo 7 en la zona central de Veracruz [Tesis de Maestría, Maestría en Antropología]. Ciudad de México: Universidad Nacional Autónoma de México; 2023. [citado 27/09/2023]. Disponible en: https://ru.dgb.unam.mx/bitstream/20.500.14330/TES01000835546/3/0835546.pdf&ved=2ahUKEwir27DfkcyBAxWQMlkFHZ46DpAQFnoECAkQAQ&usg=AOvVaw2pNzvCp-2g2Kgyi6l6RDfZ
11. Goswami R, Bello AI, Bean J, Costanzo KM, Omer B, Cornelio-Parra D, Odah R, Ahluwalia A, Allan SK, Nguyen N, Shores T, Aziz NA and Mohan RD. The Molecular Basis of Spinocerebellar Ataxia Type 7. Frontiers in Neuroscience. 2022 [citado 27/09/2023]; 16:818757. Disponible en: https://www.frontiersin.org/articles/103389/.3389/fnins.2022.818757/pdf
12. Ouchi H, Ishiguro H, Shibano K, Hara K, Sugawara M, Enomoto K, Miyata H. Primary degeneration of oculomotor, motor, and somatosensory systems and auditory and visual pathways in spinocerebellar ataxia type 7: A clinicopathological study in a Japanese autopsy case. Neuropathology. 2022 [citado 27/09/2023]; 43(2):164-175. Disponible en: https://onlinelibrary.wiley.com/doi/full/10.1111/neup.12869
13. Zahra Bouzid F, Mnasouri M, Abdelaziz C, Louhab N, Bernard S, Strubi-Vuillaume I. Spinocerebellar ataxia Type 7: clinical and genetic study of a new Moroccan family (case report). Pan African Medical Journal. 2021 [citado 27/09/2023]; 38(162). Disponible en: https://www.panafrican-med-journal.com//content/article/38/162/full
CONFLICTO DE INTERESES
El autor declara que no existen conflictos de intereses.
FUENTES DE FINANCIACIÓN
No se recibió financiación para el desarrollo del presente artículo.