CASE PRESENTATION
Parinaud syndrome due to pineal vascular tumor in a pediatric patient: a case report
Síndrome de Parinaud por tumor vascular pineal en paciente pediátrico: a propósito de un caso
Laura Isabel Moreno Miña 1*, https://orcid.org/0000-0003- 2949-1387
Lissette Zuler Miña Oliveros1,https://orcid.org/0000-0002-4730-9895
Vivian Suárez Herrera 1, https://orcid.org/0000-0002-6571-2194
Jaykel Martínez Pujol 1, https://orcid.org/0000-0002-7653-5672
1 "Faustino Pérez" Provincial Clinical-Surgical Teaching Hospital. Matanzas.
* Corresponding author: laurimoreno02@gmail.com
Received: 20/02/2026
Accepted: 21/04/2026
Published: 23/04/2026
How to cite this article: Moreno-Miña LI, Miña-Oliveros LZ, Suárez-Herrera V, Martínez-Pujol J. Parinaud syndrome due to pineal vascular tumor in a pediatric patient: a case report. MedEst. [Internet]. 2026 [cited access date]; 6:e535. Available in: https://revmedest.sld.cu/index.php/medest/article/view/535
ABSTRACT
Introduction: Parinaud's syndrome, caused by a dorsal midbrain lesion, results in supranuclear palsy of vertical gaze. In pediatrics, pineal tumors are the main cause; vascular lesions are rare and pose a challenge to management due to high surgical risk. Endocrine manifestations such as precocious puberty are often underreported despite their diagnostic value.
Objective: to present a case of Parinaud's syndrome due to a vascular tumor of the pineal gland in a pediatric patient.
Case presentation: a 10-year-old female patient presented with limitation of upward vertical gaze since age 6, precocious puberty (menarche at age 8), and inversion of the sleep-wake cycle since childhood. Ophthalmological examination confirmed supranuclear palsy of elevation, convergence-retraction nystagmus, and pupillary light-near dissociation. Magnetic resonance imaging identified a lobulated nodular lesion in the pineal gland (29 × 28 × 27 mm) with characteristics suggestive of a vascular tumor, without associated hydrocephalus. Given the high surgical risk and the four-year symptomatic stability, expectant management with active surveillance was chosen. After 18 months of follow-up, the patient remains clinically and radiologically stable.
Conclusions: Parinaud syndrome can present insidiously with incomitant vertical strabismus. Endocrine manifestations, such as precocious puberty, often precede oculomotor signs, suggesting a pineal etiology. In pineal vascular tumors with high surgical risk, expectant management with active surveillance is valid if symptoms are stable, although it requires indefinite follow-up due to the risk of bleeding.
Keywords: Parinaud'sSyndrome; Vascular Tumor of the Pineal Gland; Precocious Puberty; Watchful Waiting.
RESUMEN
Introducción: El síndrome de Parinaud, por lesión mesencefálica dorsal, causa parálisis supranuclear de la mirada vertical. En pediatría, tumores pineales son la causa principal; lesiones vasculares son raras y desafían el manejo por alto riesgo quirúrgico. Manifestaciones endocrinas como pubertad precoz suelen subregistrarse pese a su utilidad diagnóstica.
Objetivo: Presentar un caso de síndrome de Parinaud por tumor vascular de la glándula pineal en una paciente pediátrica.
Presentación del caso: Paciente femenina de 10 años con limitación de la mirada vertical hacia arriba desde los 6 años de edad, pubertad precoz (menarquia a los 8 años) e inversión del ritmo sueño-vigilia desde la infancia. El examen oftalmológico confirmó parálisis supranuclear de la elevación, nistagmo de convergencia-retracción y disociación luz-cerca pupilar. La resonancia magnética identificó lesión nodular lobulada en la glándula pineal (29 × 28 × 27 mm) con características sugestivas de tumor vascular, sin hidrocefalia asociada. Dado el alto riesgo quirúrgico y la estabilidad sintomática de cuatro años previos, se decidió manejo expectante con vigilancia activa. Tras 18 meses de seguimiento, la paciente mantiene estabilidad clínica y radiológica.
Conclusiones: El síndrome de Parinaud puede debutar insidiosamente con estrabismo vertical incomitante. Las manifestaciones endocrinas, como pubertad precoz, suelen preceder a los signos oculomotores, orientando a etiología pineal. En tumores vasculares pineales de alto riesgo quirúrgico, el manejo expectante con vigilancia activa es válido si los síntomas son estables, aunque requiere seguimiento indefinido por riesgo hemorrágico.
Palabras clave: Síndrome de Parinaud; Tumor Vascular de Glándula Pineal; Pubertad Precoz; Manejo Expectante.
INTRODUCTION
Parinaud's syndrome, also called dorsal midbrain syndrome, pretectal syndrome, or Koerber-Salus-Elschnig syndrome, is a relatively uncommon neuro-ophthalmological disorder. It is characterized by the classic triad of supranuclear palsy of upward gaze, convergence-retraction nystagmus, and pupillary light-near dissociation (1). Originally described by French ophthalmologist Henri Parinaud in 1886, this syndrome results from lesions affecting the dorsal midbrain structures, particularly the superior colliculus, the posterior commissure, and the oculomotor nuclei (2).
The pathophysiology of the syndrome is based on compression or direct involvement of the vertical gaze control centers located in the brainstem. The paralysis of upward gaze originates from damage to the vertical gaze center (rostral interstitial nucleus of the medial longitudinal fasciculus and the interstitial nucleus of Cajal). Meanwhile, the light-near dissociation is attributed to a dorsal midbrain lesion that interrupts the pupillary light reflex pathway due to compression of the dorsal interneurons of the pretectal nuclei, but it spares the more ventral pathway for the near pupillary reflex. This produces the dissociation of pupillary reflexes (3). Bilateral eyelid retraction (Collier's sign) and convergence-retraction nystagmus are due to the loss of supranuclear inhibition on the oculomotor nerve nucleus (1).
From an etiological point of view, causes of Parinaud syndrome include pineal region tumors, midbrain infarcts, hemorrhages, multiple sclerosis, arteriovenous malformations, infectious processes, and obstructive hydrocephalus (4). The distribution of causes varies significantly according to the patient's age: while neoplastic etiologies (particularly germ cell tumors and pineocytomas) predominate in children and young adults, vascular causes such as infarcts and hemorrhages are more frequent in the elderly population (1, 4). Up to approximately 65% of cases are due to primary midbrain lesions, while near 30% result from compression by pineal gland tumors (4).
The pineal gland, an epiphyseal structure located in the midline above the midbrain tectum, performs fundamental neuroendocrine functions through the synthesis of melatonin (5). This hormone, derived from serotonin, modulates circadian sleep-wake rhythms and has inhibitory effects on sexual development (6). Compression or destruction of the pineal gland by expansive processes can cause uncommon but clinically relevant endocrinological alterations, such as inversion of the sleep-wake cycle and precocious puberty, manifestations frequently underreported in the medical literature (5, 7).
Vascular tumors of the pineal region — including cavernomas, arteriovenous malformations, and hemangiomas — represent an exceptional etiology, particularly in the pediatric age group where germ cell tumors constitute 50-70% of pineal neoplasms (8, 9). Pineal cavernomas are especially rare in children, with few cases reported in the international literature (6). These angiographically occult lesions, composed of dilated sinusoidal channels lined by endothelium without a smooth muscle layer, have an estimated annual bleeding risk of 2.4% per patient-year (10).
The therapeutic management of symptomatic pineal vascular lesions depends on the histology, location, surgical accessibility, and bleeding risk (11). For patients with asymptomatic lesions or stable symptoms without a history of bleeding, expectant management with active surveillance through periodic magnetic resonance imaging is a valid alternative, especially when surgery carries a high risk of neurological morbidity (10, 12). In lesions that are difficult to access surgically, conservative observation with clinical-radiological follow-up has shown results comparable to other therapeutic modalities in terms of functional preservation (11).
The objective of the following report is to present a case of Parinaud syndrome due to a vascular tumor of the pineal gland in a pediatric patient.
CASE PRESENTATION
A 10-year-old female patient, previously healthy, came to the Neuro-Ophthalmology consultation at “Faustino Pérez” Provincial Hospital in Matanzas, Cuba, due to an eye movement abnormality noticed by her mother since approximately age 6. During the directed interview, the following relevant history was obtained:
Normal psychomotor development stands out, although with slight clumsiness for fine motor activities, and adequate school performance without learning difficulties. Since early childhood, the patient has had a marked alteration of her sleep pattern, characterized by inversion of the sleep-wake cycle, with a preference for sleeping during the day and difficulty falling asleep at night. Regarding sexual development, spontaneous menarche was recorded at 8 years of age, which shows precocious puberty, accompanied by weight and height development above the expected percentile for her age.
General physical examination:
Good general condition is evident, with weight and height above the 75th percentile for her age, which means mild obesity. Regarding pubertal development, signs compatible with precocious puberty are observed, corresponding to Tanner stage III-IV for both breast development and pubic hair. The neurological examination reveals an alert and oriented patient, without apparent motor or sensory deficits.
Ophthalmological examination:
1 - Best corrected visual acuity: 20/20 in both eyes.
2 - Extraocular motility:
- Primary Gaze Position: Orthotropia.
- Downward gaze: Normal, complete.
- Upward gaze: Bilateral and symmetric limitation of eye elevation (supranuclear palsy of elevation) (Figure 1).
- Convergence-retraction nystagmus when attempting forced elevation.
3 - Pupillary reflexes: Normal sized pupils, with light-near dissociation (slow or absent response to light, preserved to accommodation).
4 - Fundus examination: No alterations in either eye. Optic discs with well-defined edges, normal shape, size, and color. Macula and retinal vessels without alterations. Retina attached.
5 - Visual field by confrontation: Normal in both eyes.
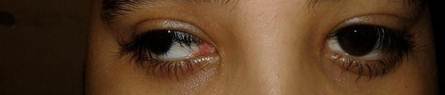
Figure 1. Limitation of upward vertical gaze. Bilateral supranuclear palsy of elevation with associated eyelid retraction is observed
Complementary studies:
Complementary studies included a brain MRI with a neuro-ophthalmological protocol, which revealed a nodular, lobulated, well-defined image in the projection of the pineal gland, measuring 29 mm × 28 mm × 27 mm. The lesion had heterogeneous signal characteristics, with areas of hyperintensity on T1 and T2 sequences suggestive of a hemorrhagic or vascular component, and showed a compressive effect on the midbrain tectum and superior colliculi. No signs of obstructive hydrocephalus or intracranial hypertension were observed in the obtained slices (Figures 2 and 3).
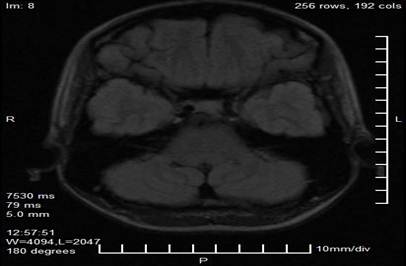
Figure 2. Axial magnetic resonance imaging cut in T1 sequence showing a lobulated nodular lesion in the projection of the pineal gland (arrow), with heterogeneous hyperintensity suggestive of a hemorrhagic or vascular component.
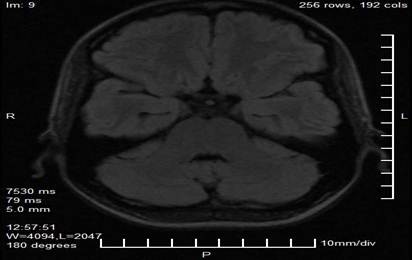
Figure 3. Axial magnetic resonance imaging cut showing the extension of the pineal lesion and its anatomical relationship with the midbrain tectum and superior colliculi (shaded area).
Presumptive diagnosis: Parinaud syndrome secondary to an expansive vascular lesion of the pineal gland.
Therapeutic approach:
The patient was referred to Neurosurgery, and a conservative therapeutic approach was decided. This decision is based on the anatomical location of the lesion in the pineal region, which is an area of difficult surgical access, on the probably vascular nature of the lesion, which carries a high surgical bleeding risk, as well as on the symptomatic stability of the patient, who has had symptoms since age 6 without evidence of progression. Additionally, the absence of hydrocephalus or signs of intracranial hypertension and the high risk of severe neurological morbidity associated with surgery in the pineal region were considered.
Consequently, an expectant management plan with active surveillance was established, which includes quarterly neuro-ophthalmological check-ups to evaluate visual acuity, ocular motility, and fundus examination, along with a brain MRI every six months to detect tumor growth or signs of bleeding. A consultation with Pediatric Endocrinology was also indicated to address precocious puberty, and general prophylactic measures were indicated, including avoiding high-risk traumatic activities and maintaining adequate blood pressure control.
Evolution and follow-up:
During the 18 months of follow-up since diagnosis, a favorable evolution has been observed that supports the initial therapeutic approach. The patient remains stable in her neuro-ophthalmological symptoms, with persistent limitation of upward vertical gaze without progression, and has not presented episodes suggestive of intracranial hemorrhage, such as severe headaches, vomiting, or altered level of consciousness. Radiologically, the pineal lesion remains stable, without significant growth on the control MRI performed at 12 months. Finally, the patient maintains a good quality of life with adequate school and family adaptation, complying with moderate restriction of physical activities as a prophylactic measure.
Informed consent: Written informed consent was obtained from the patient’s parents/guardians for the publication of this clinical case, guaranteeing the confidentiality of her identity by removing identifiable data.
DISCUSSION
This case illustrates the clinical presentation of Parinaud syndrome with an insidious evolution in a pediatric patient, whose etiology due to a vascular tumor of the pineal gland represents a rarity among the already infrequent lesions of this anatomical region. The combination of classic neuro-ophthalmological manifestations with atypical endocrinological alterations (precocious puberty and inversion of the sleep-wake cycle), together with the decision of expectant management in a lesion with high surgical risk, constitute the distinctive elements that justify the presentation of this case.
Parinaud syndrome is classically classified according to age groups and causes: pineal tumors in children and young adults, multiple sclerosis in women in their third to fourth decade, and vascular events in elderly patients (1). In the pediatric age group, germ cell tumors constitute 50-70% of pineal neoplasms, followed by pineocytomas, astrocytomas, and ependymomas (2). Vascular tumors — cavernomas, arteriovenous malformations, and hemangiomas — represent less than 5% of symptomatic pineal lesions in children (3).
The patient presents a lesion with radiological characteristics suggestive of a vascular tumor (cavernoma or vascular malformation), which is exceptional in her age group. The MRI shows the classic pattern of signal heterogeneity with hyperintense areas on T1 and T2, compatible with hemorrhages in different stages of evolution (4). Unlike germ cell tumors, which typically show homogeneous hyperintensity and contrast enhancement, vascular lesions present this "popcorn-like" pattern due to the presence of repeated microhemorrhages and organized thrombi (5).
The low incidence of pediatric pineal vascular tumors makes it difficult to establish specific management protocols. In the series of 40 cases of Parinaud syndrome reviewed by Shields et al., none corresponded to a vascular etiology in patients under 18 years of age (1). This underscores the importance of reporting these cases to enrich the available evidence.
The pineal gland, an epiphyseal structure located in the midline above the midbrain tectum, synthesizes melatonin from serotonin via the enzyme N-acetyltransferase (6). Melatonin exerts inhibitory effects on the hypothalamic-pituitary-gonadal axis, modulating gonadotropin secretion (7). Compression or destruction of the pineal gland by expansive processes interrupts this inhibition, triggering central precocious puberty (8).
In the present case, spontaneous menarche at 8 years of age, preceded by breast development and pubic hair, constitutes a classic but frequently underdiagnosed endocrinological manifestation in children with pineal lesions (9). The inversion of the sleep-wake cycle — preference for sleeping during the day and nighttime insomnia — represents a circadian alteration directly attributable to the dysfunction of melatonin synthesis (10). These symptoms, present since the patient's early childhood, preceded the evident oculomotor manifestations by years.
The medical literature has traditionally emphasized the oculomotor alterations of Parinaud syndrome, relegating endocrinological manifestations to the background (11). However, recent studies suggest that up to 30% of pediatric patients with pineal tumors present sleep disorders, and 15-20% of girls with pineal compressive lesions develop precocious puberty (12). Detailed questioning about sleep patterns and sexual development should be considered mandatory in the evaluation of children with signs suggestive of midbrain dysfunction.
Supranuclear palsy of elevation, the cardinal sign of Parinaud syndrome, results from damage to the vertical gaze center located in the dorsal midbrain. This center includes the rostral interstitial nucleus of the medial longitudinal fasciculus (riMLF) and the interstitial nucleus of Cajal, which generate the impulses for conjugate upward eye movements (13). Compression by the pineal tumor (29 × 28 × 27 mm) on the tectum and superior colliculi explains the observed dysfunction.
Convergence-retraction nystagmus, characterized by convergent retrusive saccadic movements of both eyes when attempting elevation, represents a release of primitive fixation reflexes due to cortical disconnection (14). Bilateral eyelid retraction (Collier's sign) and pupillary light-near dissociation complete the classic clinical picture, all present in this patient although with variable intensity.
It is notable that the patient maintained normal visual acuity and a normal fundus examination, despite midbrain compression. This is explained by the absence of obstructive hydrocephalus, a condition that usually accompanies large pineal tumors and causes papilledema due to intracranial hypertension (15). The presence of hydrocephalus would have radically modified the management, requiring urgent ventricular shunting.
The therapeutic approach adopted — active surveillance without direct surgical intervention — represents a complex decision that illustrates the dilemmas in pediatric neurosurgery. Vascular tumors of the pineal region carry specific surgical risks: catastrophic intraoperative hemorrhage, damage to neighboring structures (thalamus, corpus callosum, galenic veins), and severe neurological morbidity (16).
Recent evidence supports conservative management in asymptomatic lesions or those with stable symptoms. In a series of 20 patients with asymptomatic pediatric cerebral cavernomas, the annual bleeding rate was 2.4%, comparable to the adult population, and expectant management was not associated with worse functional prognosis (17). For lesions that are difficult to access surgically, such as the pineal region, observation with serial MRI constitutes a valid alternative, especially when there are no signs of intracranial hypertension (18).
However, this strategy is not without risks. The bleeding rate in symptomatic pineal cavernomas can reach 8-10% per year, significantly higher than in other locations (19). The decision for expectant management in our patient was based on: (a) documented symptomatic stability for 4 years prior to diagnosis; (b) absence of hydrocephalus; (c) radiological characteristics suggestive of a low-flow vascular lesion; and (d) high foreseeable risk of surgical morbidity.
The 18-month follow-up confirms the stability of the lesion and the absence of hemorrhagic events, validating the initial decision. However, the patient requires indefinite surveillance, given that the bleeding risk persists throughout life (20).
This case underscores the importance of systematic questioning in pediatric patients with atypical oculomotor signs. Limitation of upward gaze, although subtle, should always be evaluated using the forced elevation test in children with motor "clumsiness" or unexplained visual difficulties. The presence of sleep disturbances or accelerated sexual development in this context should alert to pineal pathology.
For the general practitioner and pediatrician, early suspicion of Parinaud syndrome allows timely referral to the specialist. The ophthalmologist, for their part, should complete the examination with evaluation of the pupils (light-near dissociation) and vertical motility, specifically looking for convergence-retraction nystagmus. MRI is the imaging study of choice, which should include specific sequences for characterizing vascular lesions (20).
This report has inherent limitations. A histopathological study confirming the vascular nature of the lesion was not available, limiting the diagnosis to radiological characteristics. MR angiography or conventional arteriography, useful for better characterizing vascular flow, were not performed due to limitations given by the unavailability of Gadolinium contrast. The 18-month follow-up, although suggestive of stability, does not allow establishing long-term prognosis.
CONCLUSIONS
Parinaud syndrome in pediatrics requires high clinical suspicion when facing limitation of upward gaze, especially if accompanied by early endocrinological manifestations (precocious puberty, sleep disorders). Pineal vascular tumors, although exceptional, should be differentiated by MRI with a heterogeneous signal suggestive of bleeding. In lesions with high surgical risk and stable symptoms, expectant management with active surveillance is a valid alternative, always with rigorous clinical, radiological, and endocrinological follow-up. The stability observed during 18 months supports this approach in selected cases.
BIBLIOGRAPHIC REFERENCES
1. Shields M, Sinkar S, Chan W, Crompton J. Parinaud syndrome: a 25-year (1991-2016) review of 40 consecutive adult cases. Acta Ophthalmol. 2017;95(8):e792-e793. doi: 10.1111/aos.13412.
2. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231-1251. doi: 10.1093/neuonc/noab106.
3. Nogueira RM, Cardoso LS, Fonseca L, Correia M, Iraneta A, Roque P, et al. Hydrocephalus in children - a rare case of pineal cavernoma and literature review. Surg Neurol Int. 2020;11:294. doi: 10.25259/SNI_231_2020.
4. Gross BA, Lin N, Du R, Day AL. The natural history of intracranial cavernous malformations. Neurosurg Focus. 2011;30(6):E24. doi: 10.3171/2011.3.FOCUS1165.
5. Ilahi S, Beriwal N, Ilahi TB. Physiology, pineal gland. StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 [cited 20/02/2026]. Available in: https://www.ncbi.nlm.nih.gov/books/NBK557791/
6. Arendt J, Aulinas A. Physiology of the pineal gland and melatonin. In: Feingold KR, Anawalt BD, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-2022. [cited 20/02/2026]. Available in: https://www.ncbi.nlm.nih.gov/books/NBK550972/
7. Cecon E, Liu L, Jockers R. Melatonin receptor structures shed new light on melatonin research. J Pineal Res. 2019;67(4):e12606. doi: 10.1111/jpi.12606.
8. Zhang Y, Sun Y, Rao J, et al. Pineal region tumors and precocious puberty: a retrospective study. World Neurosurg. 2020;143:e268-e275. doi: 10.1016/j.wneu.2020.06.292.
9. Li J, Li X, Feng D, Zhang Y, Wang Z, Liu X, et al. Clinical characteristics and surgical outcomes of pediatric pineal region tumors: a single-institution case series of 121 patients. Childs Nerv Syst. 2021;37(9):2845-2853. doi: 10.1007/s00381-021-05237-8.
10. Hardeland R. Melatonin and sleep-wake regulation: from basic science to clinical application. Sleep Med Rev. 2023;68:101783. doi: 10.1016/j.smrv.2023.101783.
11. Sheetal S, Thomas R, Byju P, Sasidharan A, Mathew F, Madhusudanan M. Parinaud's syndrome: a clinical and etiological review. Neurol India. 2024;72(4):784-790. doi: 10.4103/neurol-india.NI_270_20.
12. Jankowski PP, Vance EF, Williams K, et al. Pineal region tumors in children: a review of the literature. J Neurooncol. 2021;151(3):463-470. doi: 10.1007/s11060-021-03720-x.
13. Leigh RJ, Zee DS. The neurology of eye movements. 5th ed. New York: Oxford University Press; 2015.
14. Siatkowski RM, Zimmer B, Rojanapratikul P. Convergence-retraction nystagmus: pathophysiology and clinical implications. J Neuroophthalmol. 2021;41(2):189-195. doi: 10.1097/WNO.0000000000000987.
15. Osborn AG, Salzman KL, Barkovich AJ, et al. Diagnostic imaging: brain. 3rd ed. Philadelphia: Elsevier; 2016.
16. Palmisciano P, Ogasawara C, Nwagwu CD, Bin Alamer O, Gupta AD, Giantini-Larsen AM, et al. Pineal region metastases: a systematic review of clinical characteristics, management strategies, and survival. World Neurosurg. 2022;159:156-167.e2. doi: 10.1016/j.wneu.2022.01.005.
17. Rauschenbach L, Santos AN, Dinger TF, Darkwah Oppong M, Li Y, Tippelt S, et al. Functional outcome after pediatric cerebral cavernous malformation surgery: a single-center case series and systematic review of the literature. Sci Rep. 2023;13:29472. doi: 10.1038/s41598-023-29472-5.
18. Rauschenbach L, Bartsch P, Santos AN, Lenkeit A, Darkwah Oppong M, Wrede KH, et al. Recent novelties in research and management of cerebrospinal cavernous malformations. Acta Neurochir (Wien). 2024;166:489. doi: 10.1007/s00701-024-06378-3.
19. Horne MA, Flemming KD, Su IC, et al. Clinical course of untreated cerebral cavernous malformations: a meta-analysis of individual patient data. Lancet Neurol. 2016;15(2):166-173. doi: 10.1016/S1474-4422(15)00303-8.
20. Taslimi S, Modabbernia A, Amin-Hanjani S, Barker FG, Macdonald RL. Natural history of cavernous malformation: systematic review and meta-analysis of 25 studies. Neurology. 2016;86(21):1984-1991. doi: 10.1212/WNL.00000000000
AUTHOR CONTRIBUTION
LIMM: Conceptualization, investigation, data curation, methodology, visualization, writing of the original draft, as well as reviewing and editing the final manuscript.
LZMO: Conceptualization, investigation, and supervision.
VSH: Conceptualization, investigation, and supervision.
JMP: Conceptualization, investigation, and supervision.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
FUNDING SOURCES
The authors did not receive funding for the development of the article.
USE OF ARTIFICIAL INTELLIGENCE
The authors declare that artificial intelligence was not used in the writing of this manuscript.